
Sex Development in Humans
Section Goals
By the end of this section, you will be able to do the following:
- Recognize that both sex-linked and autosomal genes play a role in sex development in humans
- Explain how disruptions in the sex development pathway can lead to differences in sex development.
Sex differentiation during human development
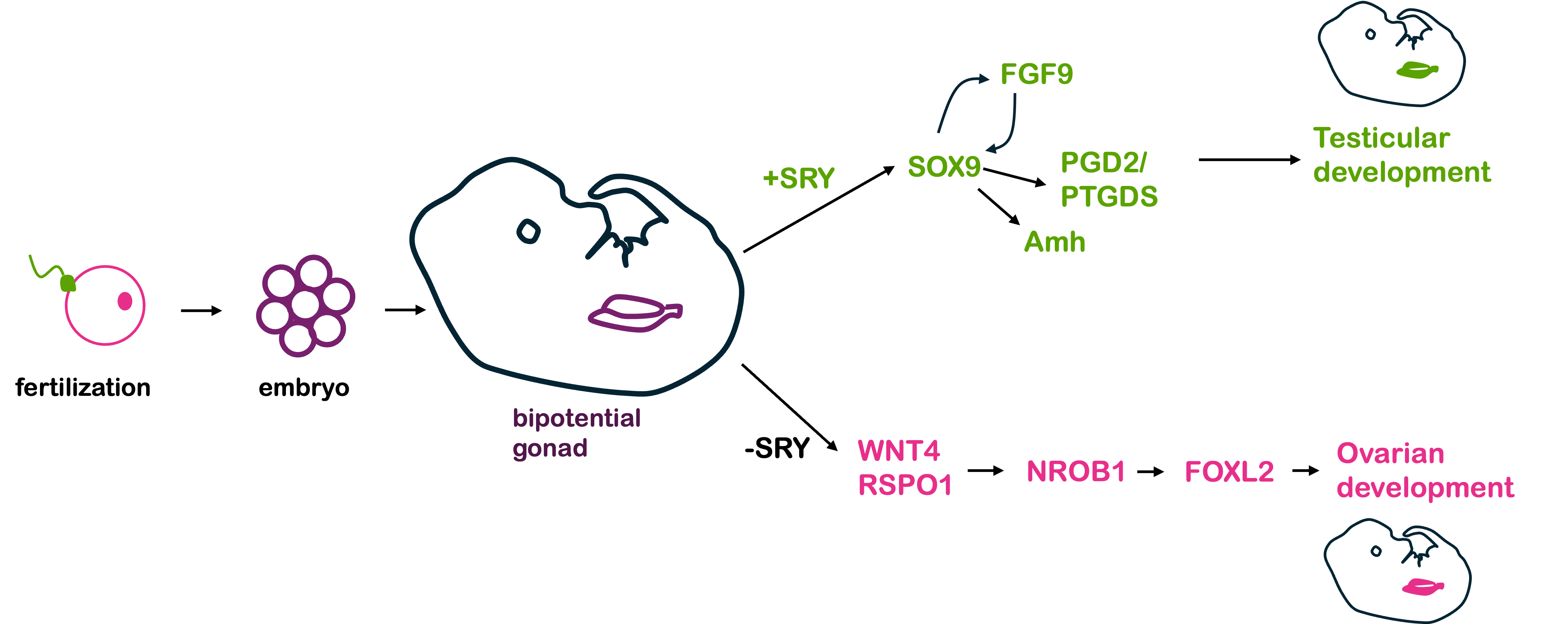
So, given what we know about genes, chromosomes, and gene expression, why does the presence of a Y chromosome typically trigger maleness during human development? The answer to that involves a network of both sex-linked and autosomal genes.
Sex is a phenotype. Typically, in most sexually dimorphic species, multiple characteristics, in addition to sex organs, distinguish male from female individuals. Those sex-associated traits are phenotypes, just like hair color, eye color, or wing shape. Those phenotypes can be genetically (or in some cases environmentally) determined.
In humans and other mammals, the Y chromosome carries the SRY gene. The SRY protein encoded by the gene is a transcription factor. In the early stages of human development, a human embryo develops a bi-potential genital ridge – that is, tissue that has the potential to become either ovaries or testes. Early embryos also have two systems of ducts, Wolffian and Müllerian, which can develop into the male and female reproductive tracts, respectively.
If a Y chromosome is present, the transcription factor SRY is produced. In turn, SRY activates the expression of Sox9, which is also a transcription factor. Sox9 in turn activates other genes, which eventually lead to the development of testes.
The testes, in turn, produce testosterone and anti-Müllerian hormone, or AMH. Testosterone (and other hormones, including 5α-dihydrotestosterone) trigger the formation of other organs in the male reproductive system from the Wolffian duct tissue, while AMH causes degeneration of the Müllerian duct and suppresses the development of female sex structures.
In the absence of SRY (as in individuals with an XX genotype), an alternative set of molecular signals is typically activated, including WNT4, RSPO1, DHH (Desert Hedgehog), and β-catenin. These lead to the development of ovaries. The ovaries then produce estrogen and trigger the development of the uterus, oviducts, and cervix from the Müllerian duct.
The sequential activation of these genes is illustrated in Figure 1, below.
Thus, in humans and other mammals, the SRY protein product sets into motion this cascade of events that leads to maleness. However, SRY is not the only gene involved in this process – and, in fact, all the other genes shown in Figure 1 are autosomal, not sex-linked! A loss of function in any of the autosomal genes will disrupt the sex determination pathway, leading to differences in sex development, discussed in greater detail below.
Differences in sex development in humans
Although the term “biological sex” is used quite often, this term is an oversimplification of the biology underlying the development of sex characteristics. There are a number of ways males and females differ from one another. As a result, there are many ways “biological sex” might be defined.
In this module, we’ve so far been focused on chromosomal sex, where an XY individual is considered male and an XX individual is considered female. But sex is usually determined or assigned at birth based on the visible presence of a penis or a vulva in a newborn. Sex can be determined by genitalia even before birth, when genitals are clearly visible by ultrasound.
Genitalia define anatomical sex. This is also sometimes called phenotypic sex, although this is an oversimplification of the term phenotype. Gonadal sex refers to the presence of either testes or ovaries. Although individuals show a range of hormonal levels, human males and females tend to have different ranges of androgens (males typically have more, females typically have less) and estrogens (females typically have more, males typically have less). This is hormonal sex.
At puberty, both males and females develop secondary sex characteristics. In males, this includes deepening of the voice, growth of facial and body hair, and broadening of the shoulders. In females, secondary sex characteristics include the growth of breast tissue, widening of hips, and onset of menses. These secondary sex characteristics typically (though not always) develop according to hormonal sex.
You might expect that these ways of defining biological sex (chromosomal, anatomical, gonadal, and hormonal) all align: XY individuals would have a penis, have testes, have relatively high levels of androgens like testosterone and low levels of estrogens, and develop male secondary sex characteristics at puberty. But this is not always true: it is possible to have an XY genotype but have female genitalia, gonads, and secondary sex characteristics. It is possible to have an XX genotype and have male genitalia, gonads, and secondary sex characteristics. It is possible to have female external genitalia and internal testes. It is possible to have genitalia with both male and female characteristics, or indeterminate characteristics. In fact, it is possible to have almost any combination of chromosomal, genital, gonadal, and hormonal sex.
Genetically, these sex phenotypes are due to genotypic changes in any one of the networks of genes responsible for sex determination and differentiation – and because there are so many genes involved in the process, there are many Differences in Sex Development (DSDs) in the human population. Another term for DSD is intersex. Some examples of DSDs in humans are listed in the section below, although this is not an exhaustive list.
Click the arrow below to read the next section
Examples of DSDs and Resulting Phenotypes in Humans
Sex chromosome aneuploidy. Remember that aneuploidy refers to an atypical number of chromosomes. They often result from nondisjunction during meiosis or mitosis: either homologous pairs or sister chromatids fail to separate during anaphase. Aneuploidies of autosomes are relatively rare in the human population, since additional copies – or too few copies – of most chromosomes would have lethal effects on a developing embryo. (Some exceptions are Trisomy 21 – Down syndrome, Trisomy 13, and Trisomy 18, all of which have significant phenotypic effects and are linked with shortened lifespan.) Aneuploidies of sex chromosomes, however, are by far more common. This is likely because the Y chromosome has very few genes – none required for life – and any extra copies of the X chromosome can be inactivated (discussed more in the next section).
Sex chromosome aneuploidies are the most common DSDs in the human population as well as the most common aneuploidies in the human population. Humans with sex chromosome aneuploidies usually have a normal lifespan but may be infertile, may have neurological and/or cognitive disabilities, or they may have few or no measurable phenotypic differences. It’s estimated that 50-75% of people with sex chromosome abnormalities never even know they have a sex chromosome aneuploidy.
Individuals without a Y chromosome typically – but not always – show a female phenotype, regardless of the total number of sex chromosomes. This is because the SRY gene on the Y chromosome is needed to initiate the development of male reproductive structures. Individuals with one or more Y chromosomes typically – but not always – show a male phenotype since the presence of SRY triggers the development of male reproductive structures during embryogenesis.
A selected list of human sex chromosome abnormalities is listed in Table 1, below, with associated phenotype.
| Genotype | Phenotype |
| XO (One sex chromosome only) | Turner syndrome. Anatomical females present with symptoms that vary in severity but may include short stature, ovarian failure, cardiac defects, and/or infertility that may be corrected with fertility treatment. |
| XXY | Klinefelter syndrome. Anatomical males present with signs and symptoms that vary in severity but may include taller than average stature, weak bones, delayed puberty, decreased muscle mass, and low sex drive. Some may go undiagnosed. |
| XYY | XYY syndrome. Anatomical males with normal fertility and sexual development. May be taller than average, have an increased risk of cystic acne, ADHD, and, to a lesser extent, autism spectrum disorder. May go undiagnosed. |
| XXX | Triple X syndrome. Anatomical females who are generally taller than average, some with subtle physical differences including wide-spaced eyes. Some patients may have learning disabilities and medical problems including infertility due to premature ovarian failure, but others may have mild or no symptoms and go undiagnosed. |
Translocation of SRY. In some anatomical males with an XX genotype, the SRY gene has been translocated to one of the X chromosomes or an autosome. This is often the result of an aberrant recombination event – crossing over between the X and Y chromosomes (or the Y and an autosome) during meiosis in the father. Such individuals appear phenotypically male but may have small testes or a urethra opening on the underside of the penis. Affected individuals may be shorter than average, require hormone treatments at puberty to trigger development of secondary sex characteristics, and may be infertile, but are otherwise healthy.
Swiyer syndrome. XY individuals who are phenotypically female, with functional vagina, uterus, and fallopian tubes but lacking ovaries. Because they lack ovaries, women with Swiyer syndrome are usually diagnosed in adolescence when they do not undergo a first period. Secondary sex characteristics do not develop without hormonal supplementation, but women can become pregnant with donated embryos. Women with Swiyer syndrome often have a deletion on the Y chromosome that inactivates the SRY gene, but the syndrome is also associated with mutations in the gene NROB1 on the X chromosome and several autosomal genes as well.
Complete androgen insensitivity (CAIS). 46, XY female phenotype. Mutations in the androgen receptor gene prevent cells of a developing embryo from responding to androgens, including testosterone. Although people with CAIS have normal-appearing female external genitalia and secondary sex characteristics, they have internal, undescended testes, and usually have a shortened vagina and lack a uterus.
Congenital adrenal hyperplasia (CAH). Mutations in the enzyme 21-hydroxylase block one step of cortisol synthesis. This in turn leads to a build-up of testosterone since cortisol and testosterone share parts of their biosynthetic pathway. This causes masculinization of the external genitalia in XX individuals, who may have ambiguous genitalia or even the appearance of normal male external genitalia. Such XX individuals have female gonads and internal genitalia.
5-alpha-reductase deficiency. These XY individuals have a loss of function mutation in the enzyme that converts testosterone to dihydrotestosterone (DHT). DHT is the hormone required for the development of external male genitalia during fetal development. Children with 5-alpha-reductase deficiency may appear to have female external genitalia or ambiguous external genitalia at birth. However, they have internal testes, and, at puberty, the testes produce much higher levels of testosterone that cause penile and scrotal tissue to grow and male secondary sex characteristics to develop. This particular DSD occurs at high frequency in certain communities in the Dominican Republic, where such people are called “guavedoces”. The term “guavedoces” very loosely translates to “penis at age twelve”, since children assumed to be female appear to grow a penis.
For an awesome infographic that breaks down DSDs and visually highlights the complexity of sex determination, see the article Beyond XX and XY: The Extraordinary Complexity of Sex Determination from the September 2017 issue of Scientific American. Click on the infographic and use the zoom tool to enlarge portions of the graphic. If you are interested in graphic design and science communication, and would like to learn more about the development of this particular infographic, check out the post Visualizing Sex as a Spectrum by graphic designer Amanda Montañez.
What is the Frequency of DSDs in the Human Population?
With an expanded understanding of biological sex, it becomes apparent that while individuals with unambiguously male or unambiguously female characteristics make up the majority of the human population, individuals frequently do not completely align with either category. Thus, although sex is often considered binary – male or female – this is not an accurate picture of human biology, since there are many ways differences in sex development can arise. Humans may be chromosomally male but phenotypically female, or vice versa. They may be chromosomally male, hormonally male, but phenotypically female. Or they may have nearly any other combination of chromosomal, anatomical, gonadal, and hormonal sex.
Estimates of how many individuals in the human population have DSDs range depending on who is counted. If only individuals with differences in external genitalia are counted, such individuals make up about 1/4500-1/2000 of the human population. However, DSDs may result in typical external genitalia but atypical alignment of chromosomal, hormonal, or gonadal sex. They may also include individuals with atypical numbers of sex chromosomes – many of whom may not even know they have such a condition. If individuals with atypical sex chromosome ploidy are included, some estimates are that individuals with DSDs make up about 2% of the human population. For comparison, this is about the same frequency as the red hair phenotype worldwide – not as uncommon as one might think. If you know people with red hair, you likely know someone with a difference in sex development, too, making sex not very binary at all.
Did I Get It?
CC Licensed Content, Shared Previously, Included in Reproduction in Populations
- Chromosomes, Genes, and Traits: An Introduction to Genetics. Author: Amanda Simons. Provided by: Pressbooks. Located at: Chromosomes, Genes, and Traits: An Introduction to Genetics – Simple Book Publishing. License: CC BY-SA 4.0
