
Cancer is caused by dysregulation of the cell cycle
Not all mutations have the potential to lead to cancer. Remember: many mutations have no phenotypic effect on either a cellular or organismal level. Other mutations may cause cell death or a cellular phenotype not associated with proliferation. For example, a somatic mutation in a gene important for cell metabolism would probably just lead to the death of the cell – it would not trigger uncontrollable growth. However, certain mutations in two types of genes do lead to inappropriate cell growth: proto-oncogenes and tumor suppressors.
Both proto-oncogenes and tumor suppressors play important roles in regulating the cell cycle.
Cell proliferation is a part of the normal physiology of a healthy organism as new cells are needed to repair injured or worn-out cells. Different tissues undergo cell division at different rates. For example, the cells lining the small intestine are normally replaced every 2-4 days, but cells of the central nervous system divide rarely, if at all, over the course of a lifetime[1]. (This is why spinal cord injuries do not heal.) However, the process of cell proliferation is tightly controlled to ensure that cells only divide when appropriate.
You’ll recall from the chapter on Meiosis and Mitosis that there are four stages to the cell cycle: G1, S, G2, and M (Figure 10). Transition from one stage to the next is tightly regulated in healthy tissue: cell division only happens under appropriate conditions. There is both positive and negative regulation of the cell cycle.
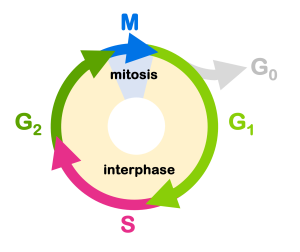
We can use the analogy of a car to describe this: the gas pedal positively regulates the car’s movement, and the brake pedal negatively regulates the car’s movement. For the car to go, the brake must be off and the gas must be on. For the cell cycle to proceed, the cell must release the negative regulation and activate the positive regulators.
Proto-oncogenes
Proto-oncogenes are positive regulators of the cell cycle. These are proteins that signal for the cell to divide – but only when conditions are right. Many of these positive regulators participate in signaling cascades that respond to growth factors. They are called “cascades” because the growth factor sets off a series of events that lead to cell division, much like a chain of dominos triggered by one push. One example of such a signaling cascade is shown in Figure 11.
In Figure 11 an extracellular signaling protein called a cytokine binds to a receptor on the cell surface, initiating a series of events inside the cell that leads to the transcription of genes involved in the immune system. This includes the proliferation of immune cells, which is necessary for an immune response. If any of the genes involved sustain a gain of function mutation, this can result in cell proliferation even in the absence of the extracellular signal.
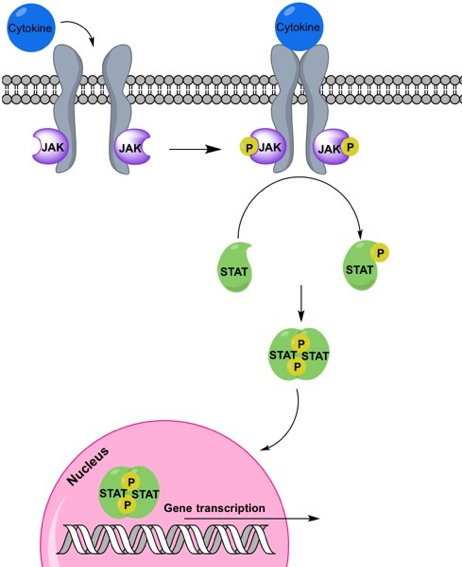
Gain of function mutations in proto-oncogenes convert the genes to oncogenes. Oncogenes cause the cell to divide regardless of whether conditions are appropriate. They’ve lost the regulatory part of their job. The family of proteins shown in the signaling cascade in Figure 11 – JAK, STAT, and many of their transcriptional targets – are mutated in many different kinds of cancers[2].
Although some oncogenic mutations can be point mutations, other larger chromosomal rearrangements are also common in the conversion of proto-oncogene to oncogene. Some of these are shown in Figure 12. For example, gene duplication can result in additional copies of a gene, which leads to extra protein production (and gain of function). A translocation can bring together a proto-oncogene with the regulatory region of another gene, resulting in the misexpression of the oncogene. A translocation can also fuse the coding sequences of two genes together, resulting in a fusion protein that is improperly regulated and overactive.
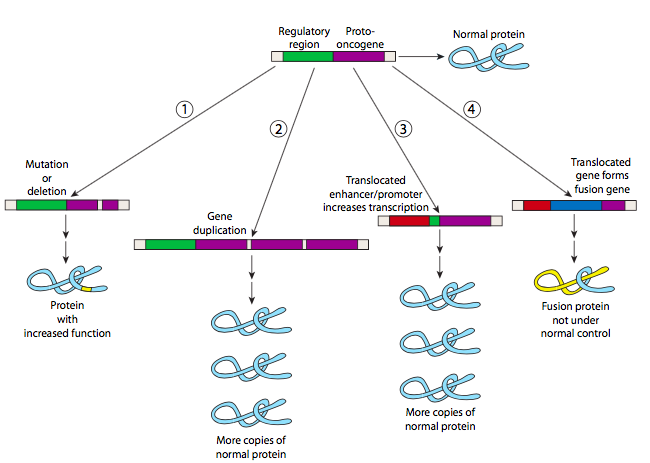
To go back to the analogy of a car, these oncogene mutations are akin to a gas pedal that is stuck to the floor.
Because these are gain-of-function mutations, they are typically dominant on a cellular level. Only one allele of a proto-oncogene needs be mutated to cause overactive growth of a cell.
The somatic gain of function mutations in proto-oncogenes is the only type of mutation in these genes that would lead to cancer. Other types of mutations in proto-oncogenes do not lead to cancer. For example, a somatic loss of function mutation in a proto-oncogene would not lead to cancer because loss of function would prevent a mutant cell from proceeding through the cell cycle. And germline mutations in proto-oncogenes are typically embryonic lethal since proper regulation of the cell cycle is necessary for a zygote to grow into an adult multicellular organism.
Tumor suppressors
If proto-oncogenes are the gas pedal of the cell cycle, the brakes are tumor suppressors. Tumor suppressor genes are involved in cell cycle checkpoints, putting the brakes on until conditions are right and ensuring the integrity of the genome.
The cell cycle is negatively regulated by checkpoint proteins that monitor conditions inside and outside the cell to be sure it is appropriate for the cell to divide. Four checkpoints are shown in Figure 13. The G1 checkpoint blocks cell cycle progression from G1 to S, monitoring for adequate cell size and nutrients as well as DNA damage. The G1 checkpoint proteins will block the transition to S phase until conditions are appropriate and any DNA damage is repaired.
The intra-S checkpoint ensures that replication is completed appropriately, since incompletely replicated DNA cannot be properly divided among daughter cells. These checkpoint proteins block cell cycle progression until replication is complete. They also monitor for DNA damage.
The G2/M checkpoint monitors for DNA damage and ensures replication is complete before the transition to M phase. And the M checkpoint monitors for spindle assembly, blocking progression if chromosomes are not properly attached to the spindle.
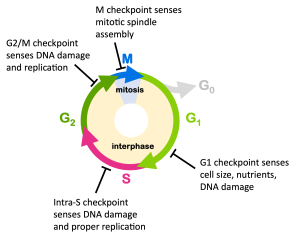
You’ll notice from Figure 13 that virtually all of the checkpoints are meant to prevent mutations: the G1, S, and G2/M checkpoints sense for DNA damage which would lead to mutations if not repaired, while the M checkpoint senses for chromosomal attachment to the spindle, which could lead to the gain or loss of entire chromosomes after cytokinesis.
Some tumor suppressors directly block the cell cycle, allowing other tumor suppressors to act in DNA repair. You can think of these two tumor suppressor functions as gatekeeper proteins and caretaker proteins. Gatekeepers block the cell from proceeding through the cell cycle, while caretaker proteins repair DNA and ensure genomic integrity.
Loss of function mutations in either type of tumor suppressor allows the cell cycle to proceed, even if DNA damage is present. These mutations are usually recessive on a cellular level; if just one healthy copy is present, this is typically enough to prevent a cell from becoming cancerous. This is shown in the cartoon in Figure 14.
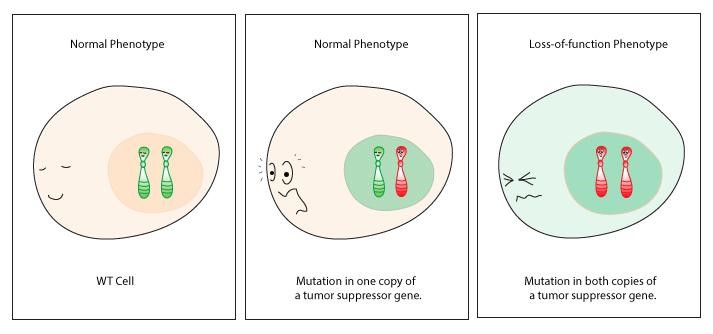
As with the proto-oncogenes, only a subset of mutations in tumor suppressor genes can lead to cancer. It typically requires loss of function mutations in both copies of a tumor suppressor for a somatic cell to become cancerous. A somatic gain of function would not lead to oncogenesis – in fact, evolutionarily, there is evidence that gain of function mutations in tumor suppressors may protect against cancer. And a germline, homozygous loss of function in a tumor suppressor is often embryonic lethal: without that function, an early embryo likely accumulates too many mutations to be compatible with life.
Media Attributions
- Figure 10 DNA Repair © Amanda Simons is licensed under a CC BY-SA (Attribution ShareAlike) license
- Figure 11 DNA Repair is licensed under a CC BY-SA (Attribution ShareAlike) license
- Figure 12 DNA Repair © Molecules and Mechanisms(Wong) is licensed under a CC0 (Creative Commons Zero) license
- Figure 13 DNA Repair © Amanda Simons is licensed under a CC BY-SA (Attribution ShareAlike) license
- Figure 14 DNA Repair © WassermanLab is licensed under a CC BY-SA (Attribution ShareAlike) license

{kind=link}