
Part II: DNA damage causes mutations
So how do these changes to DNA arise? Mutations begin with damage to DNA.
DNA damage can be endogenous (from within) or exogenous (caused by external forces). Endogenous sources of DNA damage include mistakes during replication and exposure of the DNA to certain natural byproducts of metabolism. Exogenous sources of DNA damage include exposure to UV light, carcinogens, and radiation (like X-rays). Most DNA damage is repaired by the cell, discussed in the module on Cancer Genetics. But if damage is not fixed before replication occurs again, the error can become fixed in the genome as a mutation. The next section looks at sources of DNA damage and mutation.
Replication errors can cause single base changes
Many mutations begin as replication errors, with an incorrect base inserted opposite the parent template during elongation of the daughter strand.
Figure 14. Tautomeric shift of guanine from the common keto form to the uncommon enol form.
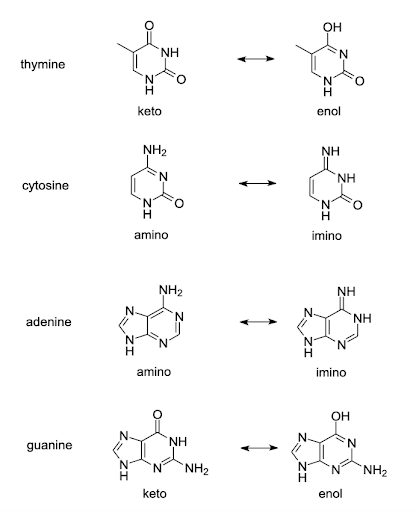
One way this is hypothesized to occur is due to the chemistry of the nucleotide bases. Although we draw the bases as static structures, the bases undergo rare tautomeric shifts, a spontaneous rearrangement of hydrogens within the structure. These changes happen spontaneously but infrequently. You can think of this almost like the bases briefly flickering back and forth from one form to another, as is shown for guanine in the animated slides in Figure 14. The changes are reversible, but the shift appears to last long enough for the replication to be affected if the replication machinery encounters the wrong tautomer[1].
Tautomeric forms of all four bases are shown in Figure 15. The common tautomers are shown left in the figure. Those are in equilibria with the less common variants shown on the right.
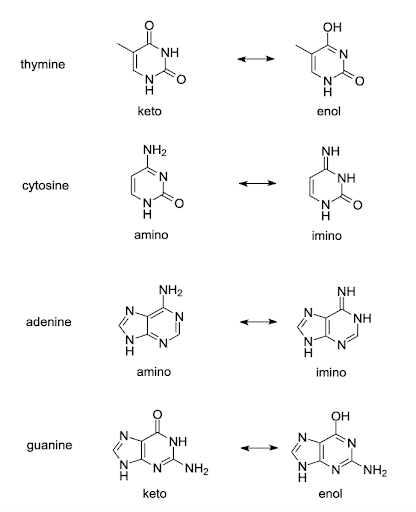
The arrangement of hydrogen bond donors and acceptors is changed in the base-pairing part of the tautomers. Although the common keto form of thymine base pairs with adenine, the rare enol form of thymine pairs with guanine. Likewise, the rare tautomers of other bases the rare enol form of guanine pairs with thymine, the rare imino form of adenine pairs with cytosine, and the rare imino form of cytosine pairs with adenine. If a base in an unwound template strand undergoes a shift, the wrong base may be incorporated in the daughter strand[2][3].
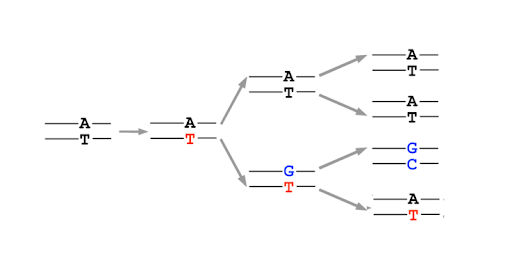
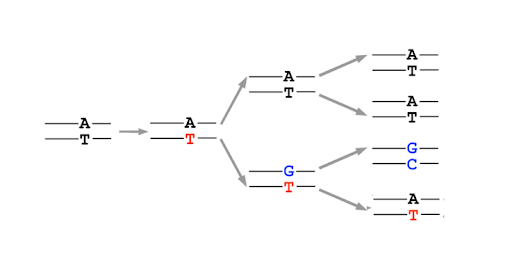
After replication, the base can shift back to the preferred amino or keto state, but this leaves behind a mismatch in the DNA. This is not a mutation yet! But it is a lesion in the DNA. Most mismatched DNA is repaired, replacing the wrong base for the right one. But if the lesion is not fixed before the next round of replication, the mis-incorporated base will be used as a template, and the daughter double helix will have a single base mutation compared to the original parent. (Figure 16)
Test Your Understanding
The types of mutations that might be caused by tautomeric shifts are called transition mutations. These are single base changes that switch a purine for a purine or a pyrimidine for a pyrimidine. Other types of DNA damage, discussed in a later section, may cause transversions, which switch a purine for a pyrimidine or vice versa. This is listed in Table 2. We will see examples of DNA damage that cause transversion mutations later in the chapter.
| Type of mutation | Base Change |
| Transition | A⬄T / purine ⬄ purine
C⬄G / pyrimidine ⬄ pyrimidine |
| Transversion | A⬄C / purine ⬄ pyrimidine
G⬄T / purine ⬄ pyrimidine T⬄A / pyrimidine ⬄ purine C⬄G / pyrimidine ⬄ purine |
Replication errors can cause insertions and deletions
Replication errors can also result in insertions and deletions of bases. This is due to strand slippage: During replication, the template and daughter strands sometimes dissociate (unpair) from one another temporarily. Replication cannot continue until the 3’ end of the growing daughter strand re-pairs with the template. But sometimes, re-pairing is misaligned, as shown in Figure 17.
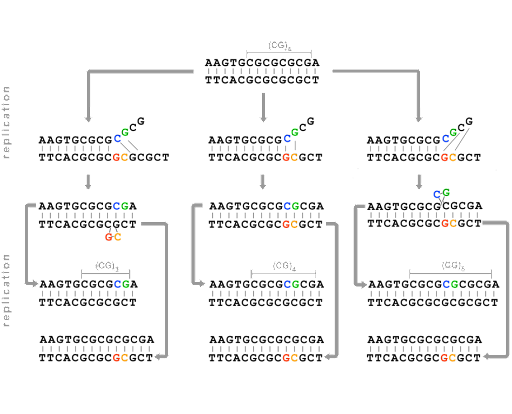
This strand-slippage happens because one or more bases are looped out when the strands re-pair. If the parent strand forms a loop, the daughter strand will be missing the looped bases, as shown on the left in Figure 17. If the daughter strand forms a loop, re-pairing the 3’ end along a segment that has already been replicated, the strand will end up with extra bases compared with the parent. This is shown on the right in Figure 17.
Strand slippage is especially common in regions of the genome with repeated sequences – like multiple CGCGCGC repeats, or triplet repeats like CAGCAGCAG. These types of repeated sequences are called microsatellites. These parts of the genome show more frequent germ-line changes than other areas of the genome, with expansion (insertion of additional repeats) or contraction (deletion of repeats) common. Because of this, they’re said to be unstable. Some studies suggest a mutation rate of about 1 in every 1000 parent-offspring transmissions, which is much higher than what is observed for the rest of the genome[4].
Many (but not all) microsatellites are in noncoding regions of the genome, and their expansion or contraction has little effect on an individual’s phenotype. Microsatellite regions in noncoding DNA do not typically affect an individual’s reproductive fitness, and microsatellite regions of the genome tend to be very variable within a population.
Differences in microsatellite length can easily be detected using PCR analysis followed by gel electrophoresis. [Although PCR is not discussed extensively here, you can see an overview of PCR from the National Human Genome Research Institute.] This made microsatellites useful in DNA fingerprinting: using DNA in forensic analysis to identify perpetrators or victims of crime, for paternity testing, or for clinical applications like tracking recovery after a bone marrow transplant. DNA fingerprinting is not limited to human populations, either. Ecologists can use DNA fingerprinting in much the same way to track familial relationships among individuals in a population.
Consequences of microsatellite expansion: Huntington’s Disease
Sometimes, though, short sequence repeats can affect phenotype. A family of genetic disorders called triplet repeat disorders results from the expansion of three nucleotide repeats. Most of the triplet repeat disorders are neurodegenerative diseases, causing neurological problems that increase in severity with age. One of the best-known is Huntington’s disease. Huntington’s disease is an autosomal dominant disorder – only one copy of the disease-associated allele is necessary to cause symptoms. Huntington’s disease causes profound neurological symptoms that appear in mid-life. These include involuntary movements, cognitive decline, behavioral problems, and dementia. These symptoms are associated with neuronal cell death. Life expectancy after diagnosis is typically around 17 years[5].
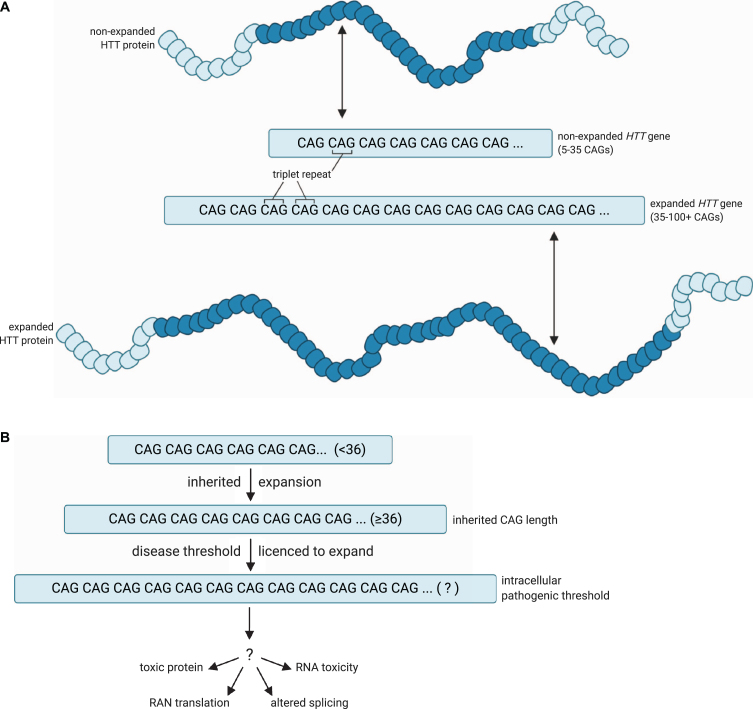
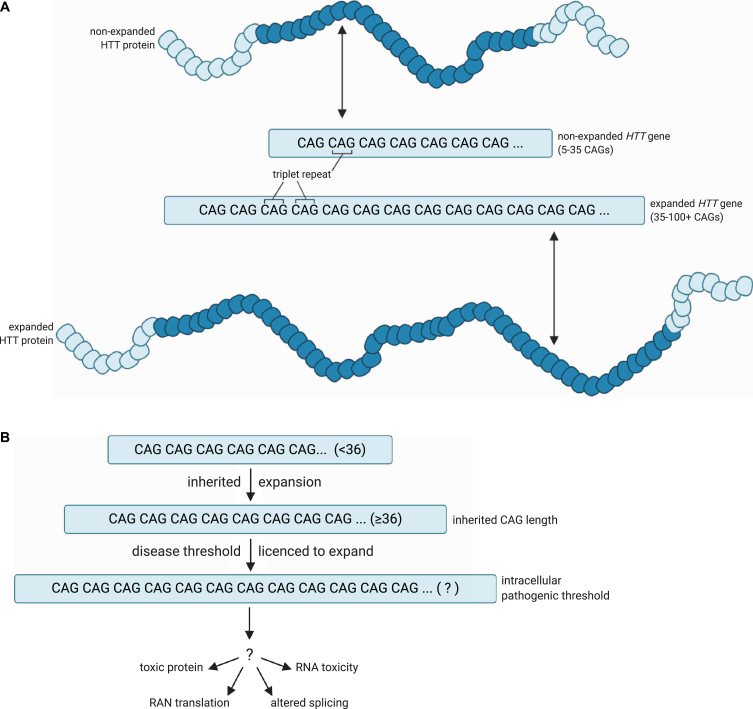
Huntington’s disease is caused by an expansion of a CAG repeat within the gene HTT, which encodes the protein huntingtin. These are insertion mutations, but they are not frameshifts since the insertion is always a multiple of three bases. CAG is the codon for glutamine, abbreviated Q. In healthy individuals, alleles have fewer than 35 CAG repeats, and the resulting protein has fewer than 35 glutamine residues in a row. Individuals with >40 repeats in at least one allele develop profound neurological symptoms between ages 30-40, with more repeats correlating with earlier onset[6].
The polyglutamine tracts tend to aggregate, or form clumps, in degenerating neurons in the brain, although it is not clear whether it is the protein or another aspect of the CAG expansion that causes neuronal toxicity. What is apparent, though, is that more than 35 CAG repeats causes the allele to be unstable through somatic cell divisions, so the repeat expands even more through multiple somatic cell divisions. The dying neuronal cells can have far more repeats than the inherited germline allele.
This is shown in Figure 18A, reprinted from Donaldson et al (2021)[7]. A healthy allele has between 5 and 35 CAG repeats and produces HTT protein with a non-expanded, non-pathogenic number of glutamine residues (dark blue in the figure). A disease-associated allele has 36-100+ CAG repeats, producing a protein with many more glutamines. In Figure 18B, the steps to disease progression are illustrated. An individual may inherit an expanded allele with 36 or more repeats that results in toxicity to those cells[8].
Repeat length is correlated with greater instability. Although people with a high healthy number of repeats (27-35) typically will not develop symptoms of H.D., their children are at greater-than-normal risk for inheriting a germline mutation with additional repeats[9]. Larger numbers of repeats are also correlated with earlier onset and increased severity of disease.
Byproducts of metabolism can damage DNA
Tautomeric mispairings and strand slippage are examples of replication errors. But in addition to replication errors, endogenous DNA damage can occur due to exposure to byproducts of metabolism, including reactive oxygen species (ROS) like hydrogen peroxide (H2O2) and superoxide (O2-). Some examples of damaged bases occurring during normal cell metabolism are shown in Figure 20.
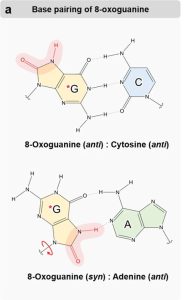
Oxidative DNA damage results in base substitutions
ROS contact can cause oxidative damage. One of the most common forms of oxidative damage is the formation of 8-oxoguanine. 8-oxoguanine can still form a normal base pair with cytosine, S, as shown at the top of Figure 19[10]. However, 8-oxoguanine can also rotate around the glycosidic bond connecting the base to the sugar, as shown in the bottom panel. If this happens, 8-oxoguanine can mispair with adenine via functional groups on the modified part of the molecule. If this mispairing occurs during replication and is not repaired, it will convert a GC base pair to a TA base pair after a second round of replication.
Test Your Understanding
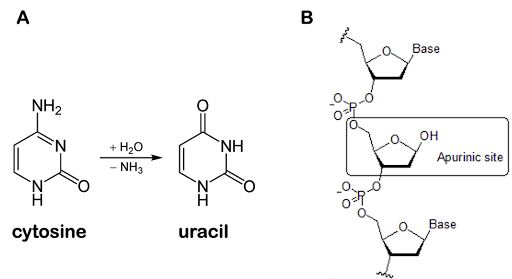
Deamination of bases and abasic sites
Other base damage that can occur includes deamination of bases and loss of bases to form an abasic site. Deamination of cytosine produces uracil, as shown in Figure 20A. The deaminated cytosine pairs with A instead of G, potentially introducing transition mutations if the damage is not corrected.
A nucleotide residue can also lose its base entirely, resulting in an abasic site, as shown in Figure 20B. Abasic sites are also called apurinic or apyrimidinic sites (abbreviated AP sites). This occurs because the glycosidic bond that connects the base to the sugar can undergo hydrolysis, a chemical reaction that breaks the bond with the addition of a water molecule. Purines are about 20 times more susceptible to hydrolysis than pyrimidines. The rate of base hydrolysis is increased when DNA is single-stranded during replication and with exposure to ROS[11].
Exogenous causes of DNA damage and mutations
Although DNA damage and a subsequent mutation can occur spontaneously or as a result of normal cellular metabolism, DNA damage can also be induced through exposure to particular kinds of chemicals. Such chemicals are called mutagens. Alkylating agents and oxidizing agents are known to cause damage to bases which is seen above. Other mutagens work differently, introducing insertions or deletions or even breaking the backbone of DNA itself.
Other causes of DNA damage include exposure to environmental radiation like X-rays or UV light. Exposure to X-rays and other forms of ionizing radiation can break the sugar-phosphate backbone of DNA, causing breaks in one or both strands of the molecule. These must be repaired before the cell replicates the DNA or undergoes mitosis or meiosis; otherwise, parts of a chromosome can be lost.
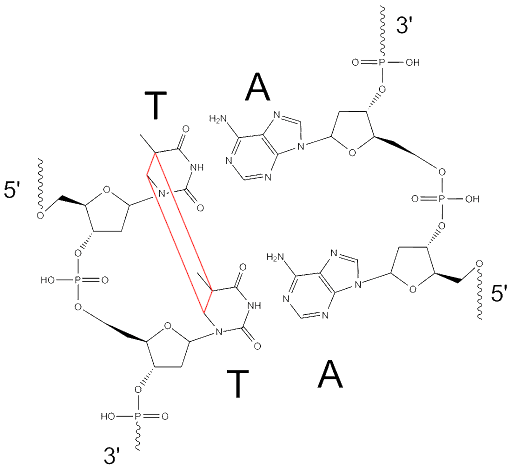
UV light causes intrastrand crosslinks, which are covalent bonds that form between adjacent pyrimidines in the backbone of DNA. They are also called pyrimidine dimers.
The structure of a thymine dimer is shown in Figure 21, but there are other ways adjacent pyrimidines can become linked as well. If not repaired before DNA is replicated, these lesions can cause frameshift mutations because the crosslinked bases may be interpreted as a single base rather than two bases.
UV light – which is part of sunlight – is a common source of somatic mutations in skin cells. This contributes to the development of many skin cancers! When you are cautioned to wear sunscreen and avoid tanning, it is because cancer-causing mutations can accumulate in skin cells over the course of a lifetime of exposure to sunlight. This is discussed more in the module on Cancer.
Cells constantly sustain damage – but most of it is repaired
You might think that DNA damage is a pretty rare event – after all, we started this chapter by saying that there is about one mutation per cell division. But, in fact, DNA damage happens far more often than that! By one estimate, in humans, around 10,000 apurinic sites (the most common DNA lesions) occur per cell every day[12]! So why are there not more mutations?
DNA damage is repaired by DNA damage response proteins. There are multiple pathways for DNA repair in cells, each of which recognizes a different form of DNA damage. Many of the DNA damage response proteins are tumor suppressor proteins, the accumulation of somatic mutations that can lead to an individual cell becoming cancerous. This is discussed more in detail in the module called DNA Repair and Cancer.
It is only when a DNA lesion escapes repair long enough to be replicated that a lesion becomes a mutation.
Media Attributions
- Tautomers © Amanda Simons is licensed under a CC BY-SA (Attribution ShareAlike) license
- Mis-incorporated base © Amanda Simons modified from Open Genetics Lectures: Original-Deyholos is licensed under a CC BY-NC (Attribution NonCommercial) license
- Strand slippage © Amanda Simons modified from Open Genetics Lectures: Original-Deyholos is licensed under a CC BY-NC (Attribution NonCommercial) license
- Pathogenic threshold in HD © Donaldson et al (2021) is licensed under a CC BY-NC (Attribution NonCommercial) license
- 8-Oxoguanine-e1708150331221-627×1024-1 © Hahm, J. Y., Park, J., Jang, E.-S. & Chi, S. W. 8-Oxoguanine: from oxidative damage to epigenetic and epitranscriptional modification. Exp. Mol. Med. 54, 1626–1642 (2022).
- Damage-cellular-metabolism © Modified from Wikipedia-Yikrazuul-PD (left) and Wikipedia-Chemist234 is licensed under a CC BY-NC (Attribution NonCommercial) license
- Thymine-dimer © Open Genetics Lectures, Original-Deyholos is licensed under a CC BY-NC (Attribution NonCommercial) license
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: *120215 COLLAGEN, TYPE V, ALPHA-1; COL5A1. last edit 05/18/2021. World Wide Web URL: https://omim.org/. ↵
- Slocombe, L., Winokan, M., Al-Khalili, J. & Sacchi, M. Proton transfer during DNA strand separation as a source of mutagenic guanine-cytosine tautomers. Commun. Chem. 5, 144 (2022). ↵
- Fedeles, B. I., Li, D. & Singh, V. Structural Insights Into Tautomeric Dynamics in Nucleic Acids and in Antiviral Nucleoside Analogs. Front. Mol. Biosci. 8, 823253 (2022). ↵
- Xu, X., Peng, M., Fang, Z. & Xu, X. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 24, 396 (2000). ↵
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: #143100: Huntington Disease. last edit 12/02/2022. World Wide Web URL: https://omim.org/. ↵
- ibid ↵
- Donaldson, J., Powell, S., Rickards, N., Holmans, P. & Jones, L. What is the Pathogenic CAG Expansion Length in Huntington’s Disease? J. Huntingt. Dis. 10, 175–202. ↵
- ibid ↵
- Migliore, S., Jankovic, J. & Squitieri, F. Genetic Counseling in Huntington’s Disease: Potential New Challenges on Horizon? Front. Neurol. 10, (2019). ↵
- Hahm, J. Y., Park, J., Jang, E.-S. & Chi, S. W. 8-Oxoguanine: from oxidative damage to epigenetic and epitranscriptional modification. Exp. Mol. Med. 54, 1626–1642 (2022). ↵
- Chastain, P. D. et al. Abasic sites preferentially form at regions undergoing DNA replication. FASEB J. 24, 3674–3680 (2010). ↵
- Thompson, P. S. & Cortez, D. New Insights into Abasic Site Repair and Tolerance. DNA Repair 90, 102866 (2020). ↵

{kind=link}
{kind=link}
{kind=link}
{kind=link}