
Epigenetics in action
The examples of epigenetic modification described above – DNA methylation and histone acetylation and methylation – are all heritable epigenetic changes that do not affect the DNA sequence. The epigenetic marks influence gene expression in a manner that is stable through cell division. The gene expression status, in turn, affects the phenotype of a cell (and the organism). The epigenetic status is maintained through mitosis, affecting the fate of somatic cells that share a parent cell. In some cases, the epigenetic status is maintained through meiosis, affecting the phenotype of offspring.
Although not discussed in this text, there are additional mechanisms of epigenetic inheritance. These include other forms of histone modification (including phosphorylation, ubiquitination, and citrullination) that contribute to the histone code. Other molecules in the cell – including non-coding RNAs – play an important role in regulating epigenetic silencing.
Epigenetics during development
Most, but not all, epigenetic marks are removed in the zygote. So as the embryo begins to develop, it starts with almost a clean slate, epigenetically. This likely contributes to the totipotency of the early embryonic cells. Totipotency describes the ability of a cell to divide and give rise to any other cell type. The cells are called totipotent stem cells. But as the fertilized egg divides, daughter cells become committed to a particular cell fate, beginning 4-5 days post-fertilization with the differentiation of trophoblast cells and embryonic stem cells of the inner cell mass (shown in yellow and red, respectively, in Figure 9). This commitment is correlated with a loss of some potential: the trophoblast cells develop into much of the placenta but cannot develop into cells of the embryo proper. Likewise, the embryonic stem cells of the inner cell mass cannot give rise to the trophoblastic structures. These embryonic cells are thus called pluripotent rather than totipotent.
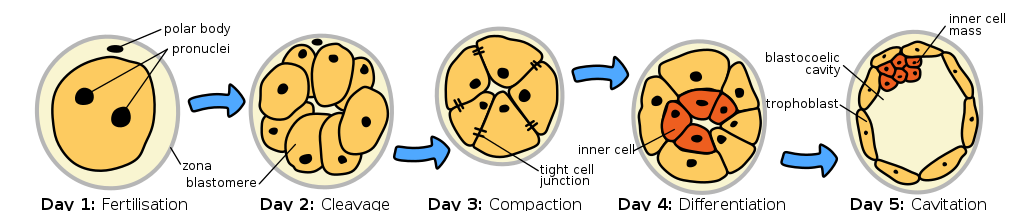
The loss of potential is associated with epigenetic silencing of certain parts of the genome[1], as are subsequent stages of differentiation. For example, genes called Nanog and Oct-4 play a role in “stemness.” Histones around these genes are acetylated in embryonic stem cells, but in trophoblast cells, the DNA around the genes is hypermethylated[2]. As cells of the inner cell mass divide and commit to a particular cell fate, additional epigenetic modifications occur.
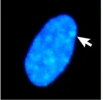
We’ve already seen an example of epigenetics in the module on Sex: X-chromosome inactivation is a form of epigenetic inheritance. In individuals with more than one X chromosome (most commonly XX genotypes, but also XXX, XXY, or other less common genotypes), only one X chromosome is typically active in each cell. The additional X chromosome(s) are highly condensed, and most (but not all) of genes are transcriptionally inactive. The condensed X chromosome is visible under the microscope as a structure called a Barr body, which is a densely staining region within the nucleus. An example of a Barr body stained is shown in Figure 10.
Which X chromosome is inactivated occurs randomly in individual cells early in embryogenesis. However, the inactivation is inherited, with the same X chromosome consistently inactive in daughter cells after cell division. X-inactivation depends on the action of a long non-coding RNA molecule called Xist. Xist molecules coat the to-be-inactivated X chromosome, recruiting additional protein factors and ultimately leading to the remodeling of the X chromatin. The inactivated X chromosome shows high levels of DNA methylation around the promoters of silenced genes as well as low levels of histone acetylation[3].
The phenotype of a calico cat is a visually striking example of how silencing of part of the genome is inherited through replication and mitosis: the patches of color result from epigenetic silencing of one or the other of the X chromosomes in a cell with XX genotype. Patches include cells that were derived from the same parent cell.
On average, because the X chromosomes are inactivated randomly, about 50% of the cells in the body of an XX individual will be inactivated for each X chromosome. But in some individuals, X inactivation is skewed, with more than 75% of cells inactivating the chromosome inherited from one parent. Interestingly, this appears to play a role in the variable expressivity of some X-linked genetic traits, including red-green colorblindness and diseases like Hemophilia B, Duchenne muscular dystrophy, and Fabry syndrome. The skewed inactivation of a healthy allele increases the severity of the disease, and the skewed inactivation of a disease-associated allele lessens the severity and reduces symptoms[4][5].
Imprinting
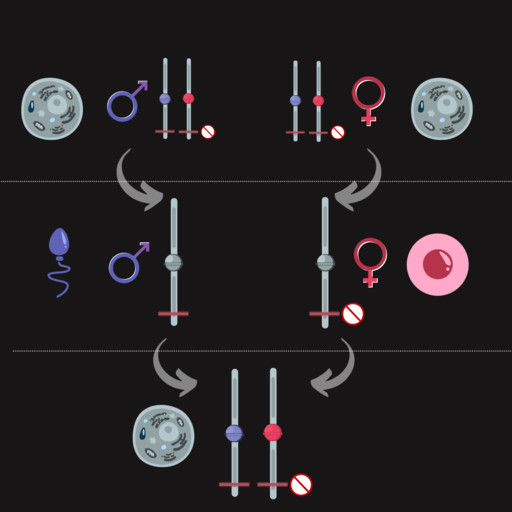
Imprinted genes are differently expressed depending on the parent-of-origin for each allele. For some imprinted genes, only (or almost only) the maternal copy of a gene is expressed, and for others, only (or almost only) the paternal copy is expressed. The other allele was silenced with DNA methylation and histone modification.
For most genes, epigenetic marks are cleared in the zygote. But for imprinted genes, epigenetic marks are reprogrammed in the egg or the sperm, and the egg- or sperm-specific epigenetic marks are maintained in the zygote beyond the stage of development when most other epigenetic marks are removed. Imprinting is maintained throughout the lifespan and is only reset during gametogenesis when new egg- or sperm-specific epigenetic marks are reprogrammed for the next generation. Figure 11 shows this for a maternally imprinted gene. Paternal imprinting is similar but with the paternal allele silenced.
The phenomenon of imprinting appears to have evolved independently in flowering plants and mammals; about 1% of human genes are imprinted. Many imprinted genes appear to be involved in growth rate, metabolism, brain function, and behavior, with imprinted genes linked to phenotypes like body size and behaviors like risk-taking and impulsive behavior[6].
In humans, several genetic diseases are caused by a disruption in imprinting. For example, Beckwith-Wiedemann syndrome is caused by a disruption in imprinted genes on Chromosome 11. These genes include the growth factor IGF2, which is normally only expressed from the paternal chromosome. Altered DNA methylation, duplication of the paternal chromosome, and other changes result in two copies of the gene being expressed. Among other signs associated with the syndrome, children with Beckwith-Wiedemann syndrome tend to be larger in size than their same-age peers[7].
Angelman syndrome is linked with a loss of function of the gene UBE3A on Chromosome 15. UBE3A is imprinted and in cells of the central nervous system, only the maternal copy of the gene is expressed. Children with Angelman syndrome show intellectual disability, small head size, and, often, a happy and excitable personality. These traits are linked with a deletion or loss of function in the maternal chromosome, which leaves patients without a functional copy of the gene in cells of the central nervous system. Mutations in the paternal allele will not cause Angelman syndrome[8].
Test Your Understanding
Epigenetic changes are influenced by behavior and environment
Over the course of a lifetime, diet, behavior, and environment contribute to the accumulation of epigenetic marks. For example, some nutrients in the diet are a source of methyl groups that are eventually part of S-adenosyl methionine (SAM/SAMe), which in turn donates methyl groups to methylate cytosine or methylate histones[9]. Nutrients like folate and choline are examples, and SAMe itself can be purchased as a dietary supplement. Other nutrients may promote or inhibit the action of DNA methyltransferase or histone modifiers. Examples include molecules like resveratrol (found in red wine, activates HDACs), curcumin (found in turmeric, inhibits HATs), and diallyl disulfide (found in garlic and onions, inhibits HDACs)[10]. Stress, drug use, trauma, and lifestyle choices have also been shown to impact the epigenome.
Environmental effects can add up over time. Identical twins, who share 100% of their DNA, are phenotypically similar but not actually indistinguishable from one another. Identical twins tend to have very similar epigenomes early in life. However, as identical twins age, their genomes stay the same, but their epigenomes diverge. This likely contributes to identical twins becoming more dissimilar as they age[11].
These environmental effects on the epigenome likely influence the incomplete penetrance of some genetic traits. For example, the methylation of a gene called REELIN appears to be linked with the development of schizophrenia. Schizophrenia is a psychiatric disorder that manifests in early adulthood. The phenotype shows a strong genetic component, but twin studies reveal that less than 50% of identical twins are concordant for schizophrenia. Differences in methylation status between twins may account for at least some of this difference[12].
Epigenetic changes are also linked with cancer. Loss-of-function mutation of tumor suppressor genes contributes to oncogenesis. However, in some cases, the DNA sequence of tumor suppressor genes is unchanged, but the gene is highly methylated, resulting in loss of function without underlying mutation. Acetylation of genes that drive the cell cycle can play a role in the uncontrolled growth associated with cancer. A number of HDAC inhibitors have been approved for the treatment of cancer[13].
Test Your Understanding
Transgenerational epigenetics
The examples discussed above all involve mitotic inheritance of epigenetic marks, with maintenance of the epigenome in somatic cells of a single individual. As was discussed earlier, most epigenetic marks are erased during gametogenesis or in the fertilized egg (zygote). However, some epigenetic marks escape this zygotic reprogramming. These allow epigenetic information to be passed through meiotic cell division from parent to offspring – a mechanism for Lamarckian inheritance of acquired traits.

There are many known examples of transgenerational epigenetic inheritance in non-mammalian species, where the epigenome can be manipulated and tracked for many generations. For example, in the plant Arabidopsis thaliana, a mustard weed used as a model organism in the genetics lab, DNA methylation induced in one generation can be measured for at least 8 generations of offspring[14].
In mice, a “tail kink” phenotype (Figure 11) is associated with a mutation of a gene called AxinFu, but whether the tail kink phenotype is penetrant depends on the methylation status of the gene. The phenotype can be inherited across generations[15].
In contrast, in humans, many of the traits suspected to be affected by epigenetics are likely multifactorial, and causative genes may or may not yet be linked with the phenotype. Many studies are retrospective analyses of human populations, tracking traits associated with environmental factors over the course of several generations.
Some of the most commonly cited of these retrospective studies track the descendants of individuals who were exposed to famine in utero. These studies include the Dutch famine winter of 1944-1945, food scarcity in the Överkalix municipality in Sweden, the Ukrainian famine of 1932-1933, and the Chinese famine of 1959-1961.
Exposure to famine as a fetus increased the risk for obesity and other metabolic disorders, Type 2 diabetes, schizophrenia, and depression. It also altered the risk of disease in their children and grandchildren[16].
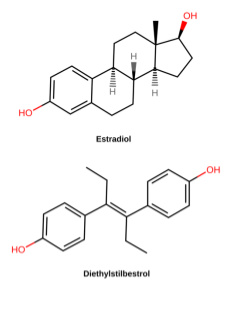
Other large-scale retrospective studies track the descendants of women treated with diethylstilbestrol (DES) during pregnancy. DES is a synthetic estrogen that was used to prevent miscarriage in pregnant women in the 1940’s-1960’s (Figure 12). By the 1970s, however, studies had shown that DES was not effective in preventing miscarriage and, even worse, was causing vaginal and breast cancers and reproductive anomalies in “DES daughters” – children born to women treated with DES. Follow-up studies have shown that DES grandchildren are also affected: DES grandchildren born to DES daughters are more likely to be born premature, and DES grandsons born to DES daughters appear to have an increased risk of certain anomalies of the reproductive tract. It is not clear whether the grandchildren born to DES sons are similarly affected[17].
Multigenerational retrospective studies like the famine and DES studies can be very powerful in linking phenotypes to environmental stressors. And, with modern genome sequencing tools, both mutations and changes to DNA methylation can be compared across generations. However, it can be difficult to distinguish between true transgenerational epigenetic inheritance and the direct effects of environmental exposure.
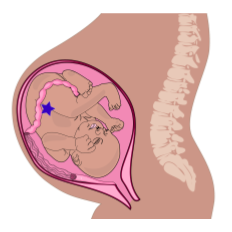
You may have heard that when a baby is born, ovaries already contain all the eggs the baby will have in their lifetime (this is not true for sperm, which are produced lifelong after puberty). Those future egg cells form very early during fetal development: as early as 8 weeks post-fertilization. So as soon as those primitive ovaries form during development, exposure during pregnancy potentially exposes three generations to a stressor: the pregnant parent (P or F0 generation), the fetus (F1 generation), and, potentially, the future eggs of the fetus (F2 generation). This is illustrated in Figure 13, which shows three generations in the same image: parent, fetus, and the eggs of the fetus, illustrated by the blue star.
Because of this, to show true transgenerational epigenetic inheritance – with the maintenance of epigenetic marks through meiosis – retrospective studies of environmental stress must track great-grandchildren from pregnant women and grandchildren from non-pregnant women and/or men.
Test Your Understanding
Media Attributions
- HumanEmbryogenesis © Wikipedia is licensed under a CC BY-SA (Attribution ShareAlike) license
- DAPI © BMC Biology via. Wikipedia is licensed under a CC BY-SA (Attribution ShareAlike) license
- Imprinting maternal © Wikipedia is licensed under a CC BY-SA (Attribution ShareAlike) license
- Cloned mice © Wikipedia is licensed under a CC BY-SA (Attribution ShareAlike) license
- Diethylstilbestrol © Medgirl131 via. Wikipedia is licensed under a CC BY-SA (Attribution ShareAlike) license
- Placenta system © Modified from Wilfredor, via. Wikipedia is licensed under a CC BY-SA (Attribution ShareAlike) license
- Xu, R., Li, C., Liu, X. & Gao, S. Insights into epigenetic patterns in mammalian early embryos. Protein Cell 12, 7–28 (2021). ↵
- Hattori, N. et al. Epigenetic regulation of Nanog gene in embryonic stem and trophoblast stem cells. Genes Cells 12, 387–396 (2007). ↵
- Fang, H., Disteche, C. M. & Berletch, J. B. X Inactivation and Escape: Epigenetic and Structural Features. Front. Cell Dev. Biol. 7, (2019). ↵
- Sun, Z., Fan, J. & Wang, Y. X-Chromosome Inactivation and Related Diseases. Genet. Res. 2022, 1391807 (2022). ↵
- Jørgensen, A. L. et al. Different patterns of X inactivation in MZ twins discordant for red-green color vision deficiency. Am. J. Hum. Genet. 51, 291–298 (1992). ↵
- Tucci, V. et al. Genomic Imprinting and Physiological Processes in Mammals. Cell 176, 952–965 (2019). ↵
- Beckwith-Wiedemann syndrome: MedlinePlus Genetics. https://medlineplus.gov/genetics/condition/beckwith-wiedemann-syndrome/. ↵
- Angelman syndrome: MedlinePlus Genetics. https://medlineplus.gov/genetics/condition/angelman-syndrome/. ↵
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 19, 3425 (2018). ↵
- Davis, C. D. & Ross, S. A. Dietary Components Impact Histone Modifications and Cancer Risk. Nutr. Rev. 65, 88–94 (2008). ↵
- Fraga, M. F. et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. 102, 10604–10609 (2005). ↵
- Imamura, A. et al. Genetic and environmental factors of schizophrenia and autism spectrum disorder: insights from twin studies. J. Neural Transm. 127, 1501–1515 (2020). ↵
- Yoon, S. & Eom, G. H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 52, 1–11 (2016). ↵
- Fitz-James, M. & Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 23, 325–341 (2022). ↵
- ibid ↵
- Vaiserman, A. & Lushchak, O. Prenatal famine exposure and adult health outcomes: an epigenetic link. Environ. Epigenetics 7, dvab013 (2021). ↵
- Zamora-León, P. Are the Effects of DES Over? A Tragic Lesson from the Past. Int. J. Environ. Res. Public. Health 18, 10309 (2021). ↵

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}