
DNA variants can be molecular markers for linkage
Linkage maps have limitations.
First, as it turns out, crossing-over does not occur uniformly across a chromosome. So-called “hotspots” for recombination exist, so a centiMorgan in one part of the genome may correspond to many more base pairs than a centiMorgan in another part of the genome. And recombination may occur at different rates in different organisms, so a centiMorgan in one species might include many more base pairs than in another. (In humans, a cM equals about 1 million base pairs[1] depending on genomic location.) Therefore, a linkage map is good for relative orientation of genes, but it does not directly translate to a physical map of DNA sequence.
Second, test crosses were most useful in organisms like fruit flies and plants, where thousands of offspring with variable traits could be measured in one controlled cross. But test crosses are not possible in all organisms or under all conditions, and not all organisms have such easily identifiable single-gene phenotypes. So, instead, in humans and other organisms, tracking the linkage of phenotypes with molecular markers was often more useful.
In the chapter on Mutation, we discuss different types of mutations and polymorphisms. Many neutral polymorphisms exist in the population of any species. Most are not within gene coding sequences or regulatory regions, although there are some exceptions. These polymorphisms are nevertheless quite useful as molecular markers, differences in DNA that can be detected molecularly and treated as codominant alleles.
Vocabulary: Just as alleles are versions of a gene, morphs are versions of a locus.
These polymorphisms, like all DNA variants, arise in any population through mutation. But because most do not affect reproductive fitness, there is no selective pressure for them to be maintained in or lost from the gene pool. As a result, there is a lot of variability among individuals in a population in certain regions of the genome. [In contrast, a mutation in part of the genome that is very important to an organism’s function is likely to negatively affect reproductive fitness and be lost from a population. Such regions of the genome are said to be highly conserved, meaning that little variation is observed.]
Some examples of variants used as molecular markers are shown in Figure 19.
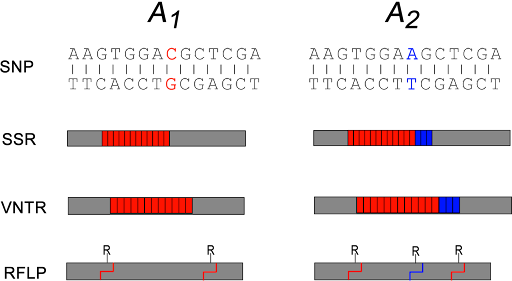
Some SNPs may change restriction sites in the genome. Restriction sites are sequences that are recognized by a restriction endonuclease which cut DNA in a sequence-specific manner. The change in sequence either creates or destroys a restriction site, affecting the length of the DNA fragment that is generated by the cut. This special subset of SNP is therefore called a restriction fragment length polymorphism, or RFLP.
SSRs are simple sequence repeats – repeating sequences like CAGCAGCAG or CCGCCGCCG. They are also called short tandem repeats (STRs) or microsatellites. VNTR stands for variable number tandem repeat. These are longer repeated sequences, where the repeated element is longer than just a few nucleotides. These sequences are usually detected by PCR of the region around the repeat, with longer repeats yielding a longer PCR product.
Detection of SNPs via PCR, RFLP analysis, and microsatellite analysis via PCR all generate different size DNA fragments that can be distinguished via gel electrophoresis. So gel electrophoresis analysis of morphs can substitute for analysis of phenotypes, with the added advantage that both alleles can usually be detected simultaneously (instead of just a dominant allele). An example of this is shown in Figure 20, where 3/15 F2 offspring are recombinant, making the recombination frequency 20%.

Using gel electrophoresis, a band on a gel can be treated as a “phenotype”.
RFLP mapping was used in 1983 to identify the first disease-associated gene: the causative gene for Huntington’s disease (HD). HD is an autosomal dominant, progressive neurological disorder with an age-of-onset in middle age. Disease symptoms include physical tremors, dyscoordination, and cognitive decline. Thoughts and mood can be affected, and many patients have hallucinations. There is no effective treatment to slow the progression of the disease, and it is ultimately fatal.
HD is caused by a triplet-repeat expansion in the HTT gene, which encodes the protein huntingtin. This is discussed further in the chapter on Mutation. To identify the gene that causes HD, several very large families with multiple members with Huntington’s disease were tested for linkage with RFLPs, which identified an RFLP on chromosome 4 that was often co-inherited with the HD phenotype[2].
Media Attributions
- DNA polymorphisms © Nickle and Barrette-Ng via. Online Open Genetics is licensed under a CC BY-NC-SA (Attribution NonCommercial ShareAlike) license
- Measure recombination frequency © Nickle and Barrette-Ng via. Online Open Genetics is licensed under a CC BY-NC-SA (Attribution NonCommercial ShareAlike) license
- Centimorgan (cM). https://www.genome.gov/genetics-glossary/Centimorgan. ↵
- Gusella, J.F., Wexler, N.S., Conneally, P.M, Naylor, S.L., Anderson, M.A., Tanzi, R.E., Watkins, P.C., Ottina, K., Wallace, M.R., Sakaguchi, A.Y., Young, A.B., Shoulson, I., Bonilla, E., Martin, J.B. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238 (1983). ↵

{kind=link}
{kind=link}